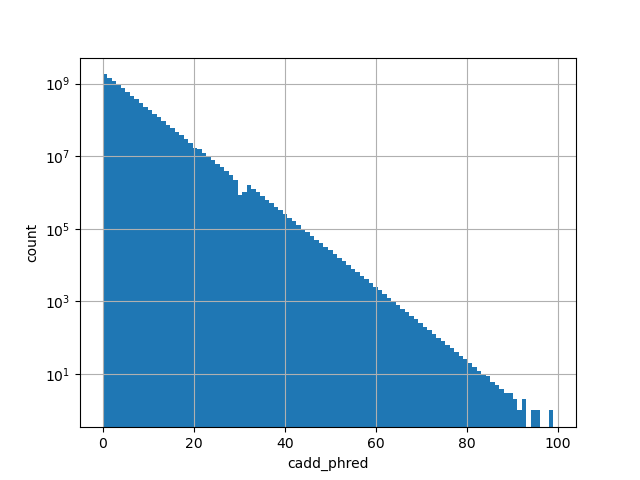
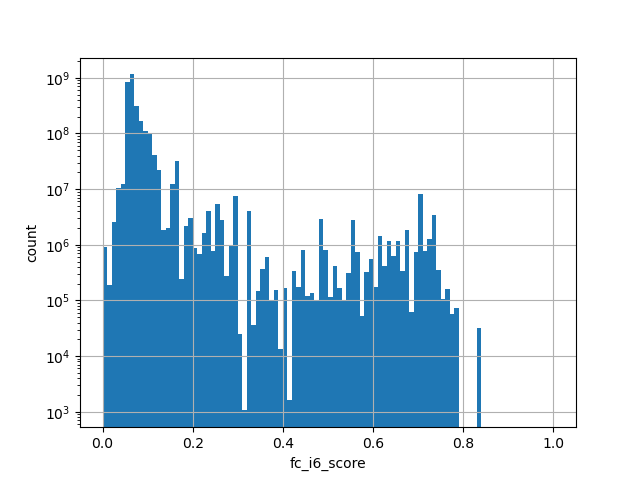
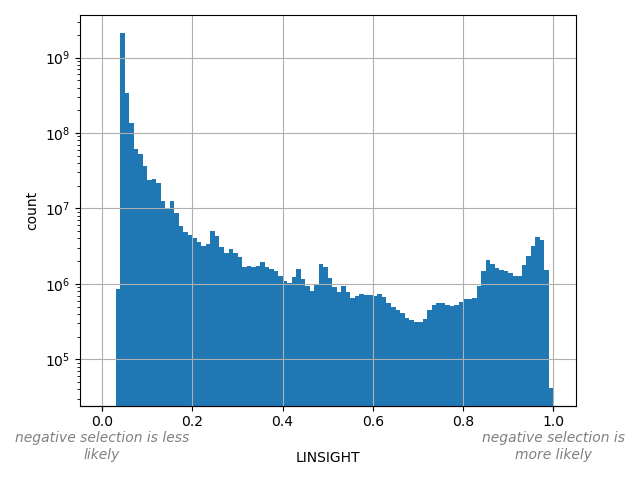
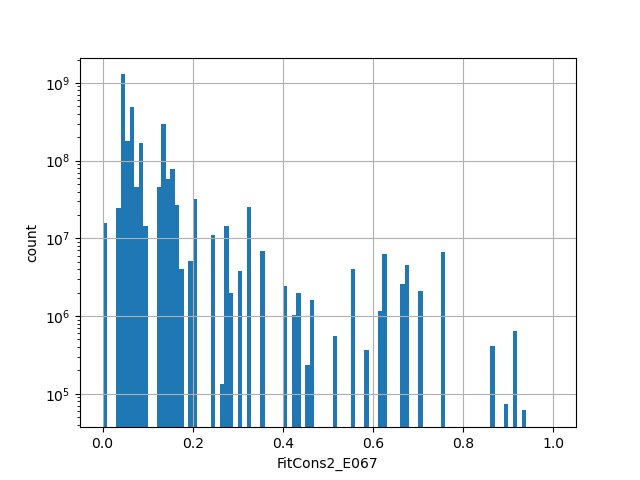
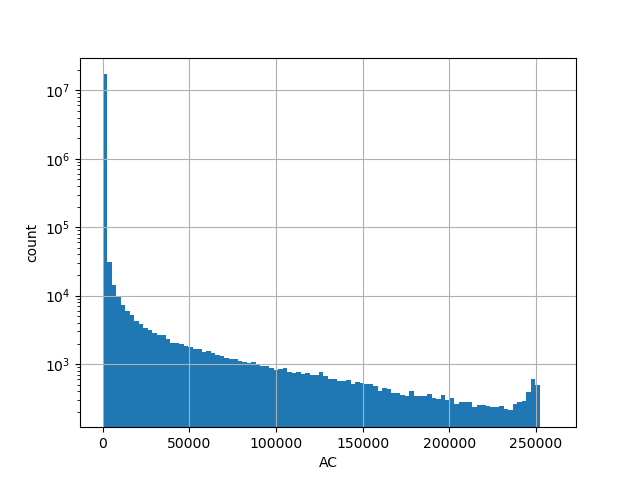
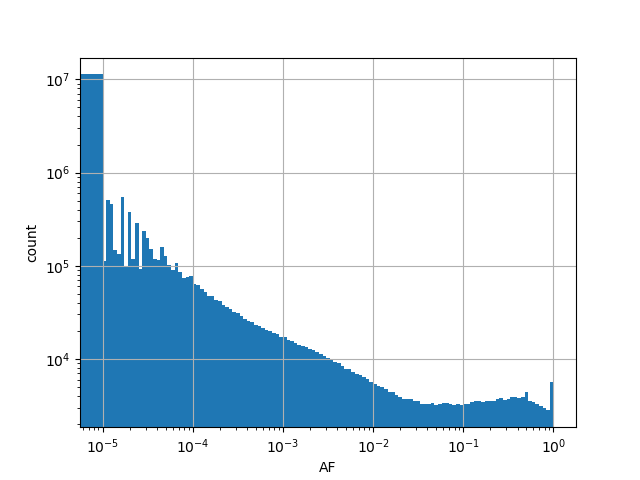
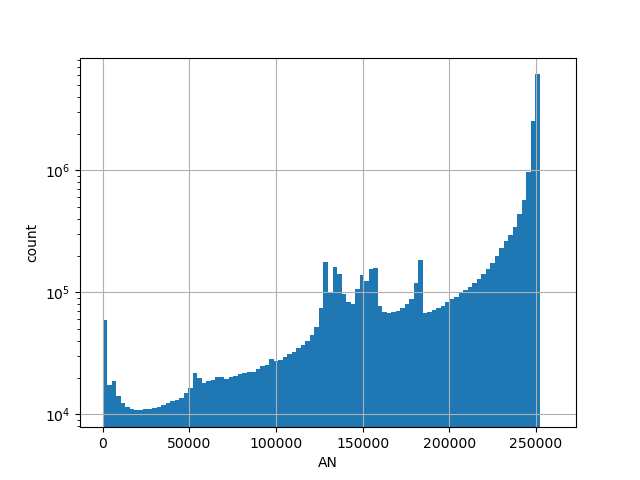
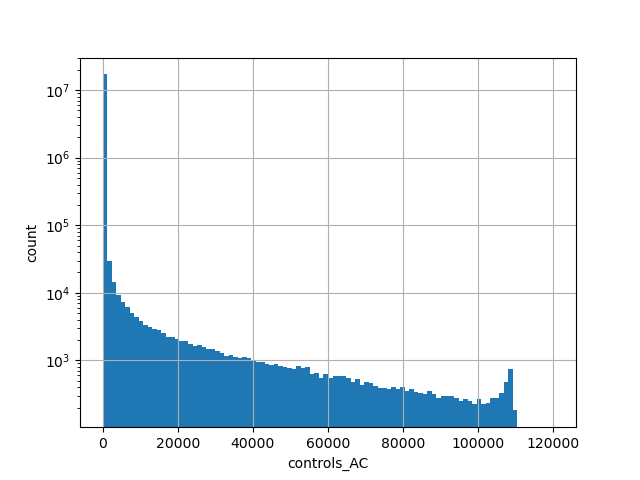
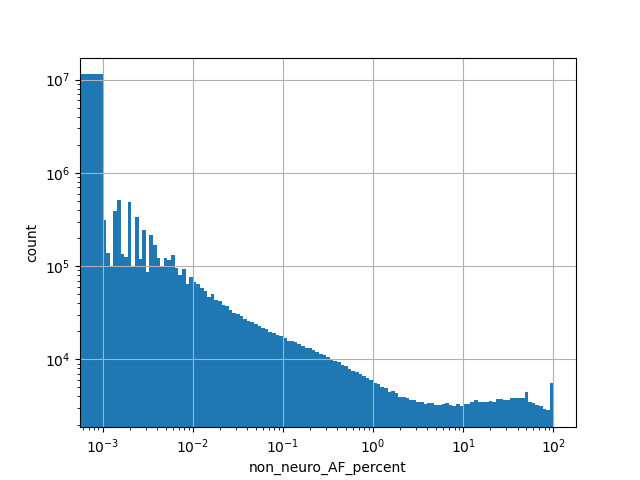
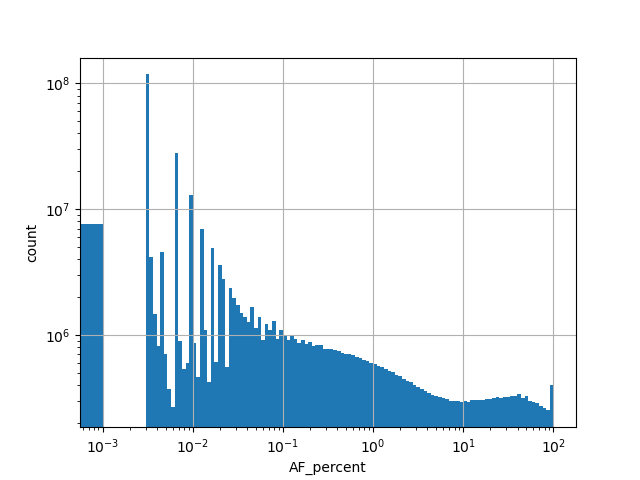
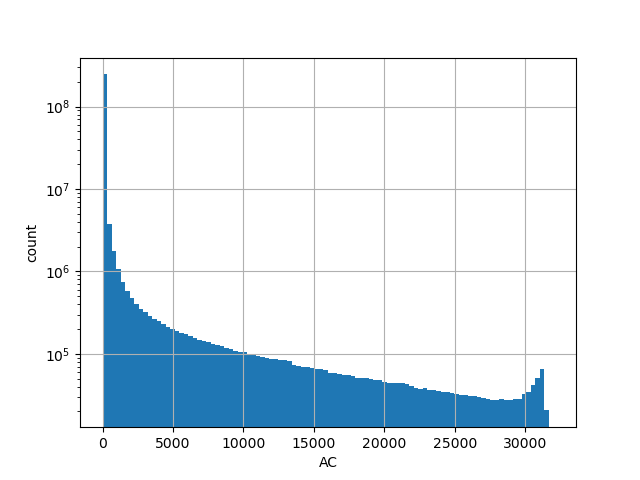
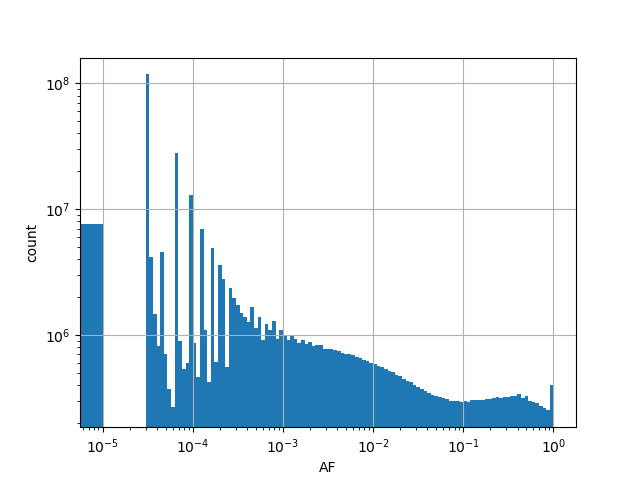
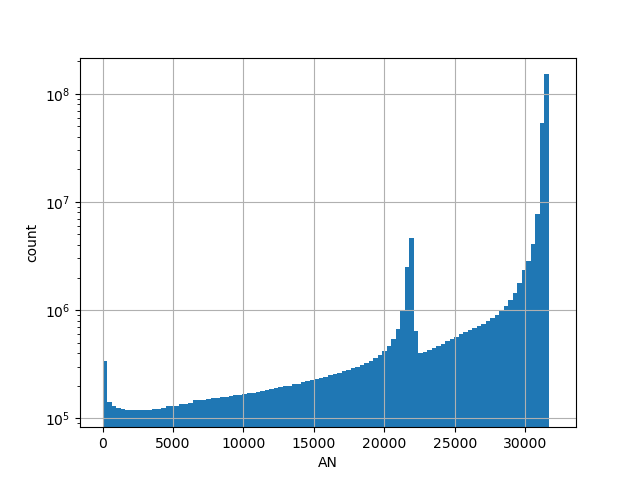
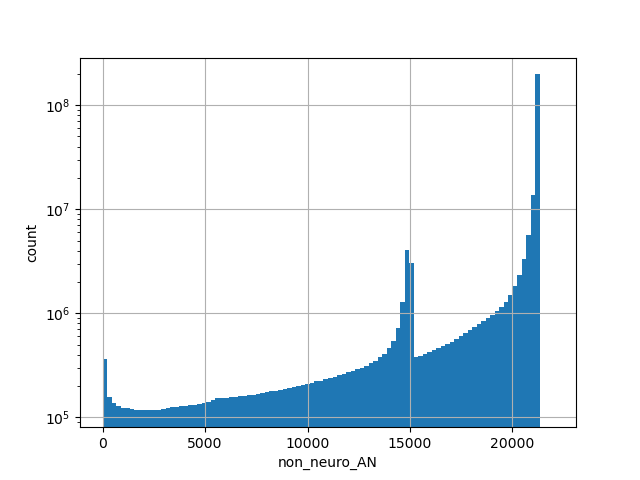
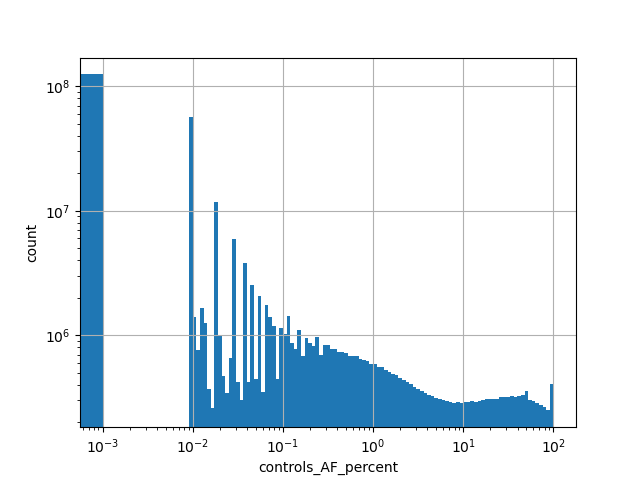
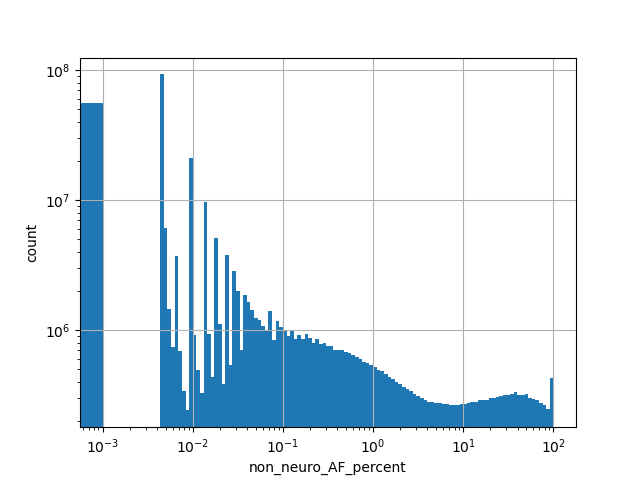
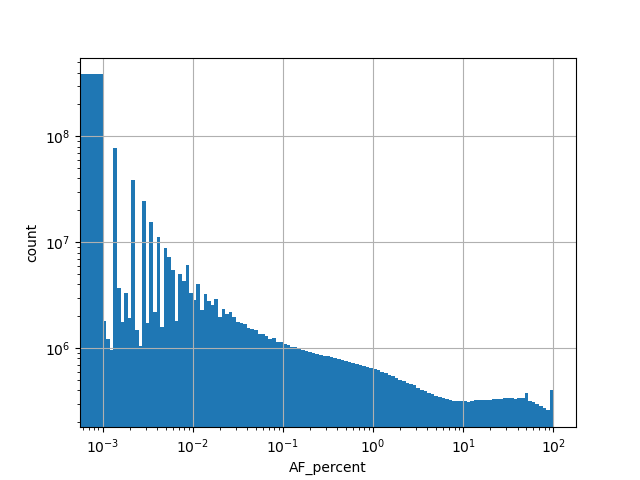
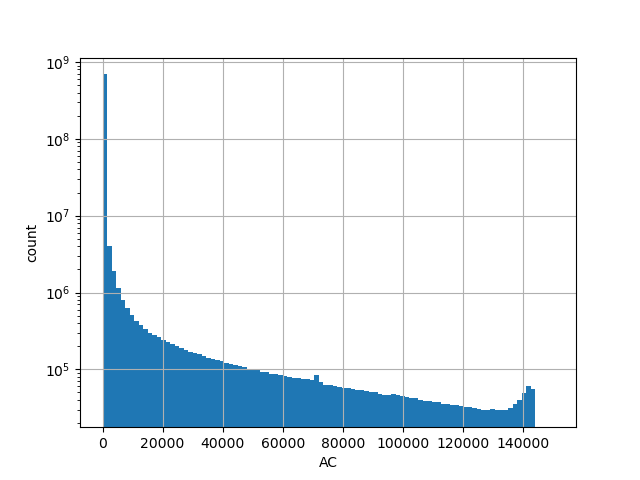
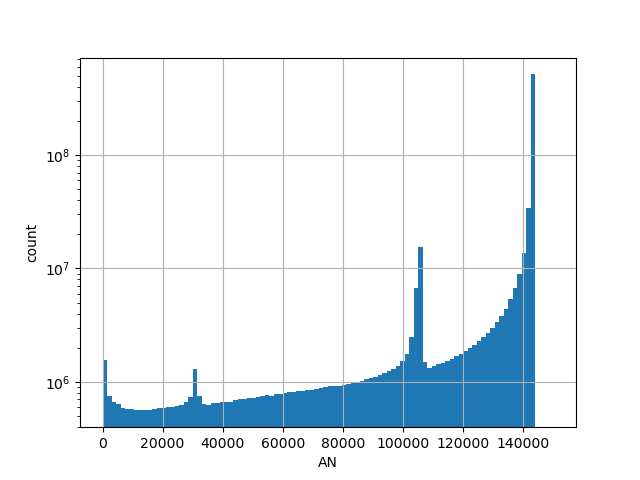
phylop100way
Type:
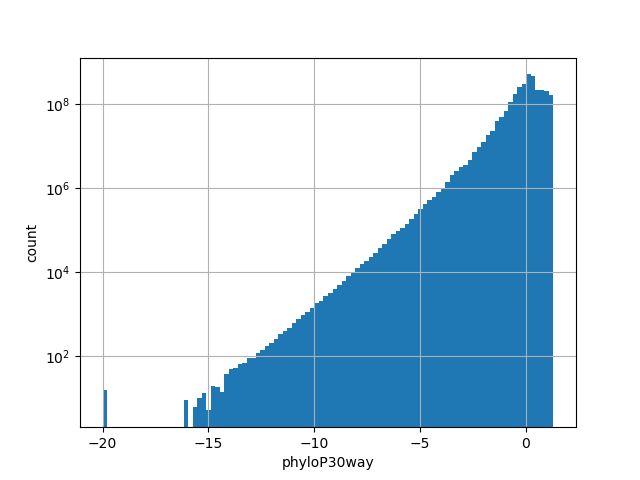
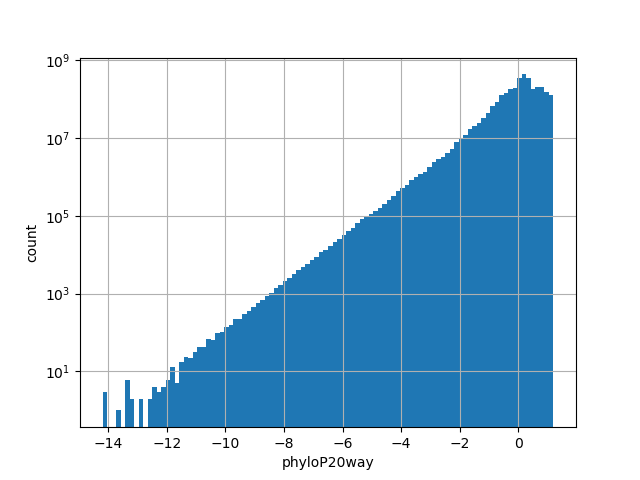
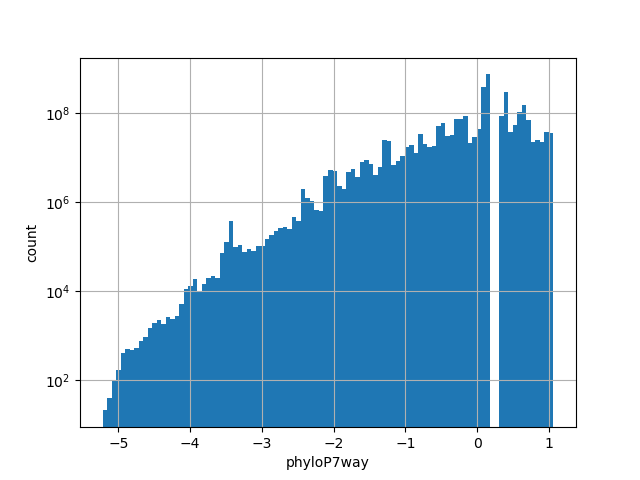
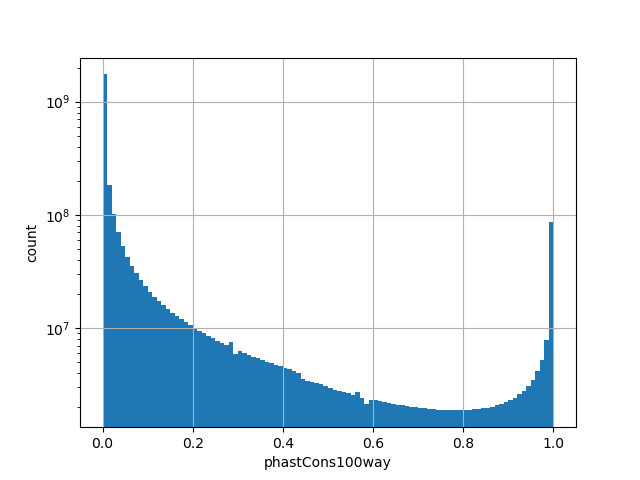
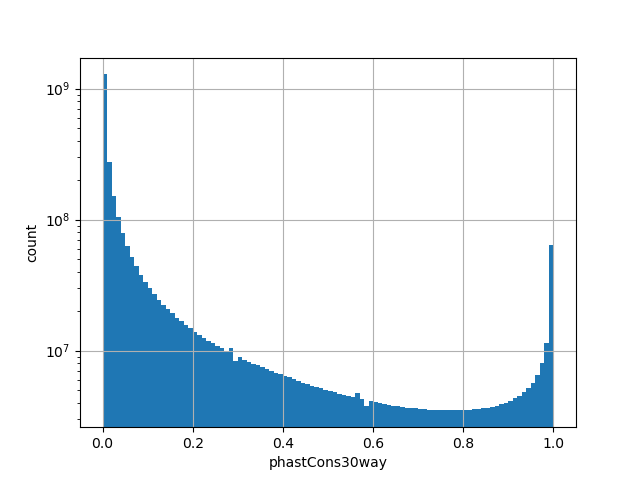
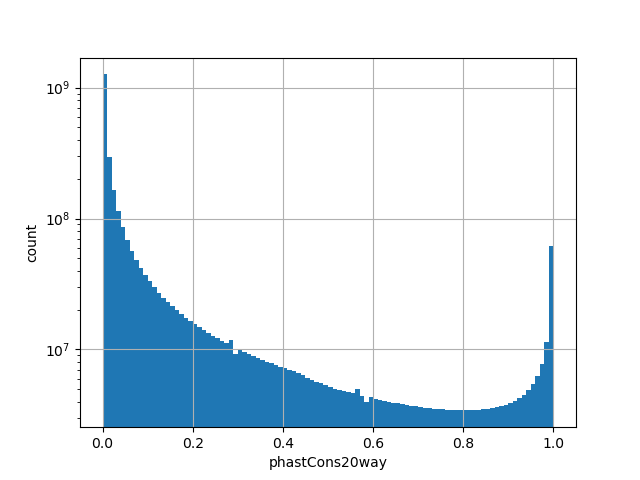
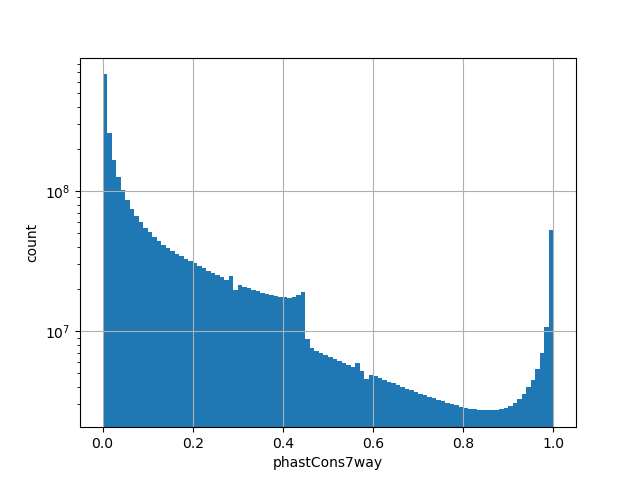
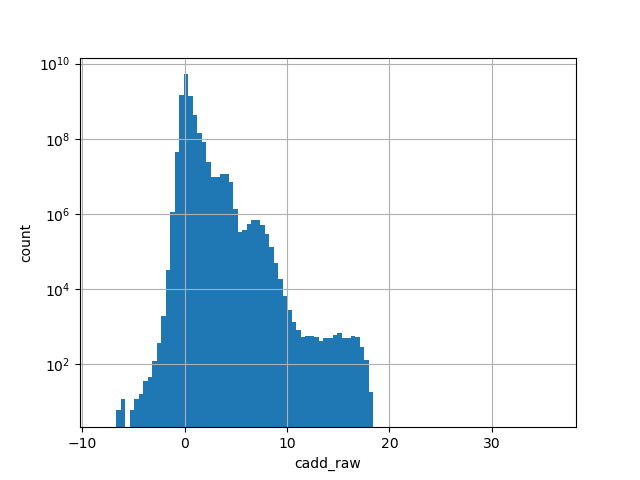
The score is a number that reflects the conservation at a position.
position_aggregator: mean [default]
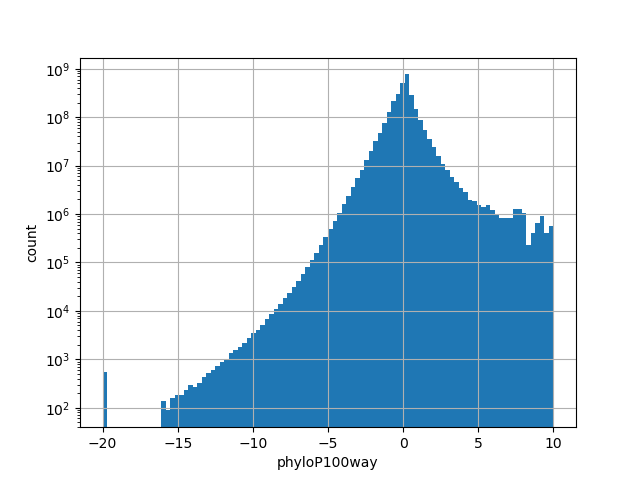
source: phyloP100way
Annotator type: position_score
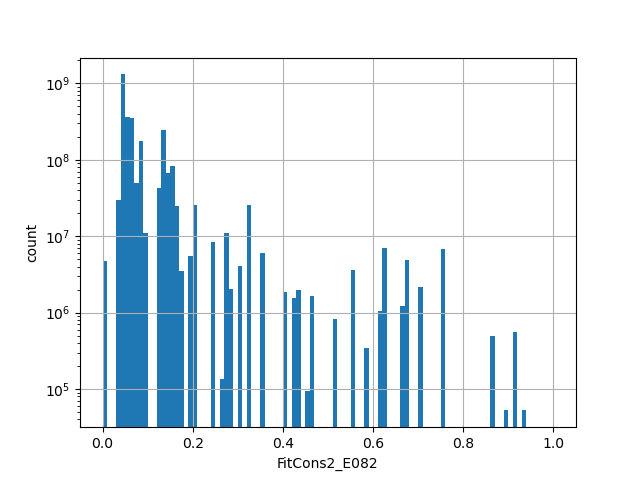
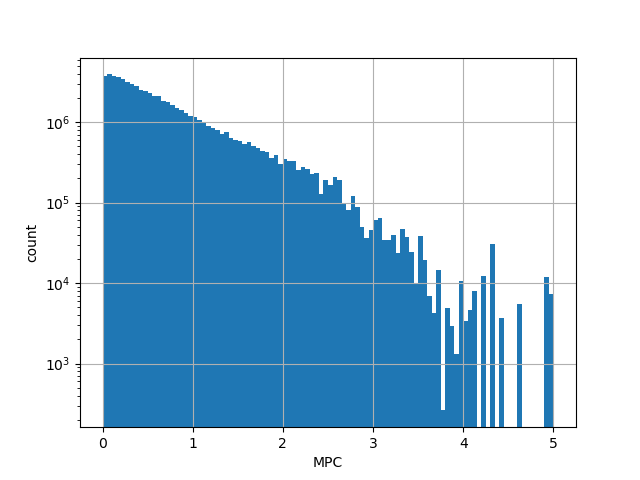
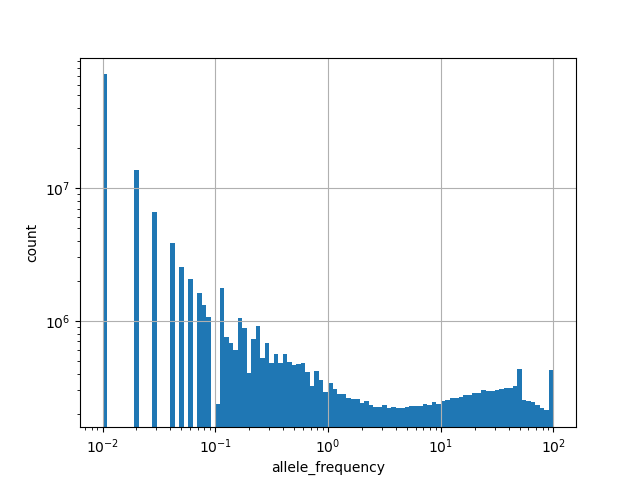
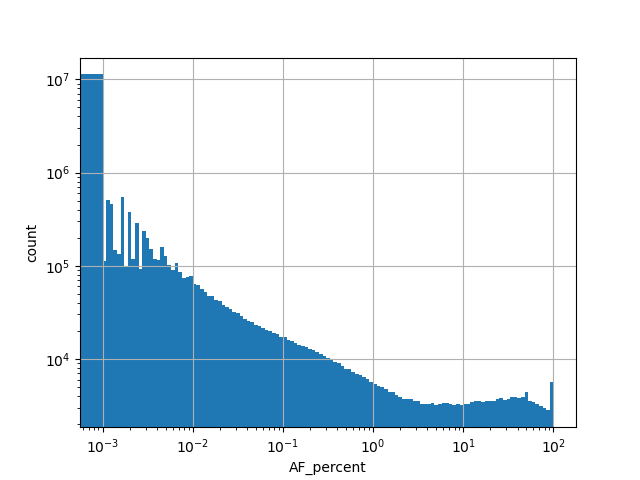
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
Resource
Type: position_score
Summary:
Conservation score based on the multiple alignment of 100 species