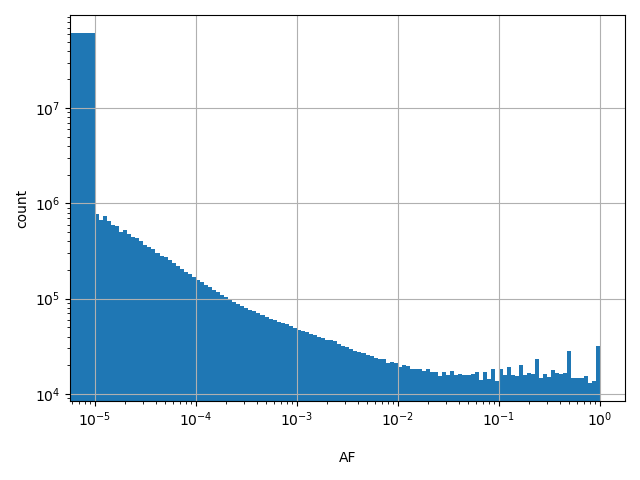
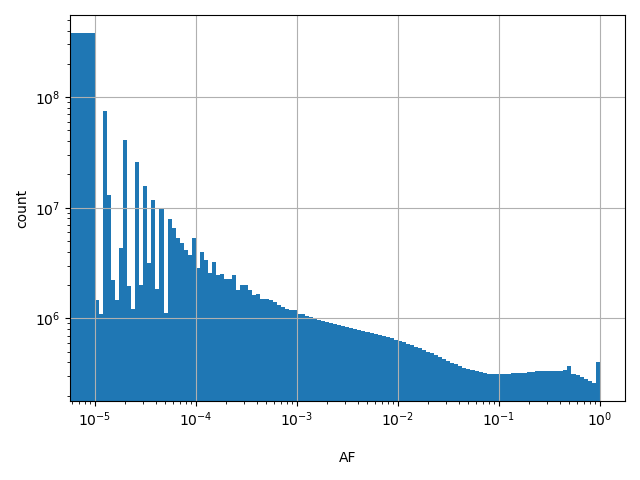
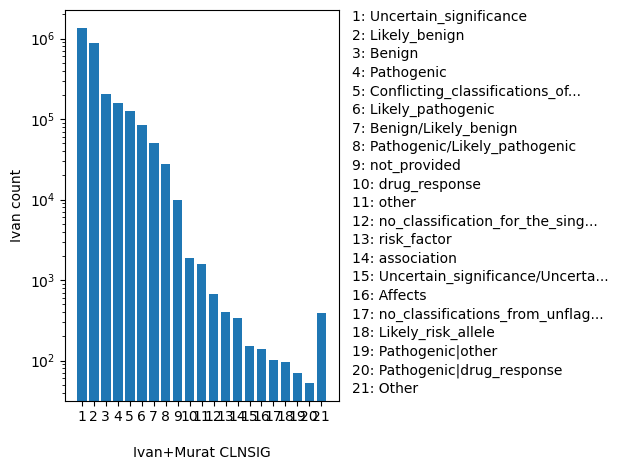
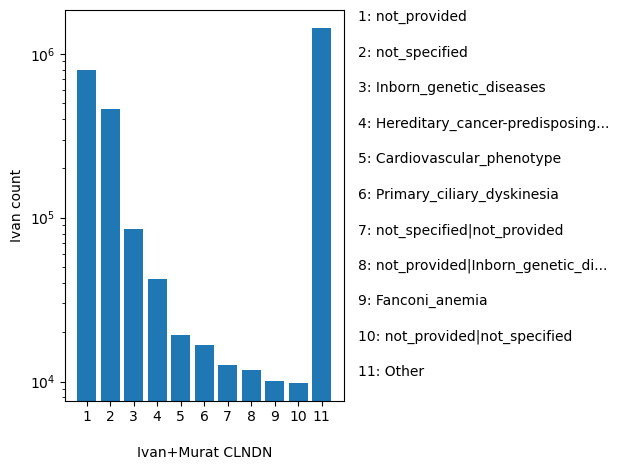
dbSNP_RS
Type:
dbSNP ID (i.e. rs number)
position_aggregator: list [default]
allele_aggregator: list [default]
source: RS
Annotator type: allele_score
Annotator to use with scores that depend on allele like variant frequencies, etc.
- input_annotatable:
normalized_allele
Resource
Type: allele_score
Summary:
dbSNP: A public database of genetic variations for research and clinical use.